Proteomics
What is proteomics?
"Proteomics is the large scale study of proteomes. A proteome is a set of proteins produced in an organism, system, or biological context." [1]
Why study proteomics?
"Proteomics is the large scale study of proteomes. A proteome is a set of proteins produced in an organism, system, or biological context." [1]
Why study proteomics?
|
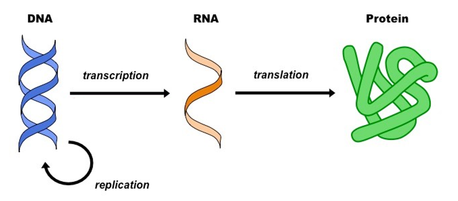
On the left is the central dogma of molecular biology. It represents how biological information changes forms in nature. When the information changes forms during the process of transcription and translation the number of potential biomolecules increases. The complexity gets compounded at each step resulting in the proteome being the -ome witht the largest number of species.
|
How is proteomics studied?
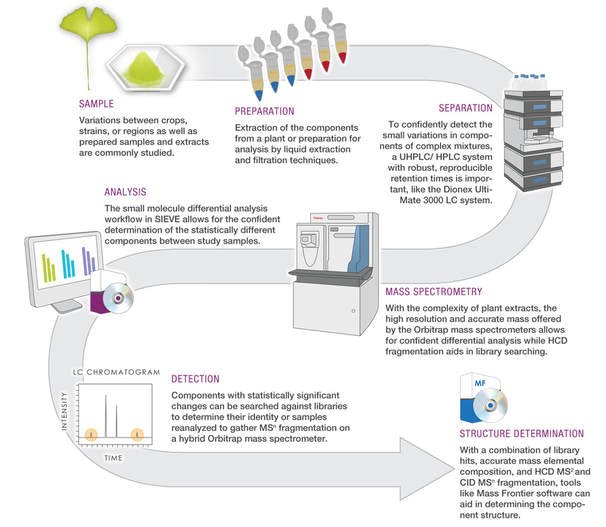
Through the use of high-throughput experiments and specifically mass spectrometry (MS).
Through the use of high-throughput experiments and specifically mass spectrometry (MS).
|
Mass spectrometers work by destroying a biomolecule and then using the fragments to elucidate what the initial biomolecule was.
For more information on MS in this video by Shomu's Biology channel on Youtube. |
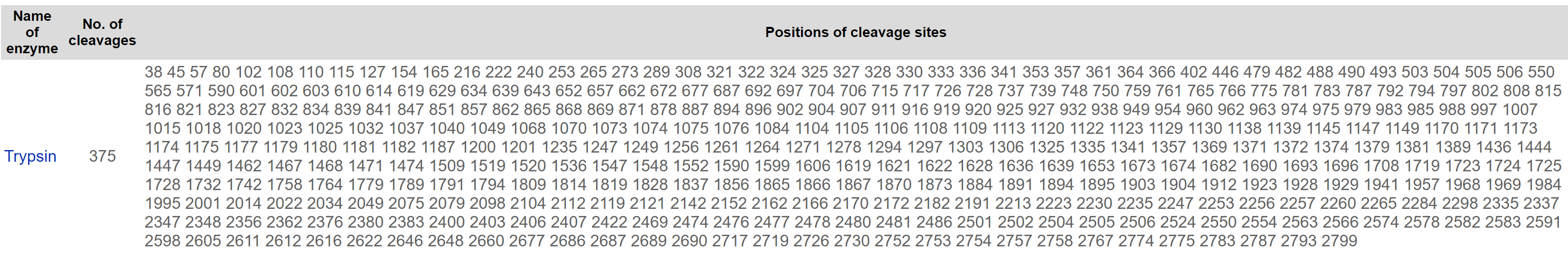
Predicted enzymatic digestion of NIPBL
During protein preparation for MS the protein is cut into smaller fragments using an enzyme. Below are the predicted cutting sites for the Trypsin enzyme when it is used to cut NIPBL. To make these predictions I used the "PeptideCutter" tool available online at the ExPASy bioinformatics resources portal through the Swiss Institute of Bioinformatics (web.expasy.org/peptide_cutter/.).
Phosphoproteomics of NIPBL
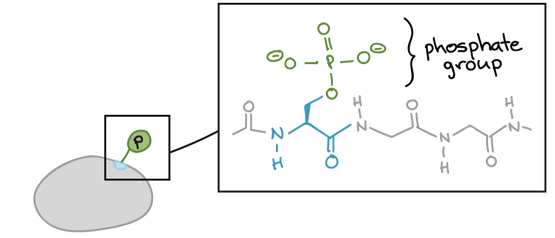
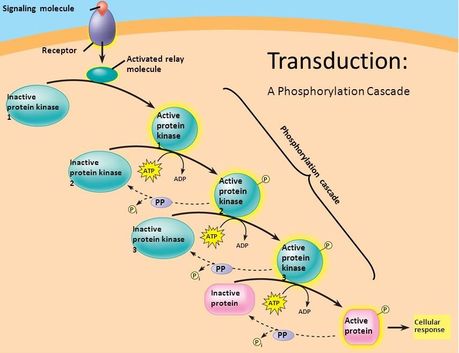
Phosphoproteomics is the study of all phosphoproteins in a biological system, including detection of phosphoproteins, locating phosphorylation site, and the quantification of differentially expressed phosphoproteins [2]. A protein becomes a phosphoprotein after it has been phosphorylated, wherein a phosphate group has been added to the protein. This post-translational modification often results in a change in function or changed signal that the modified protein is communicating to other cells [3].
Addition of phosphate group Signal cascade caused by phosphorylation
Addition of phosphate group Signal cascade caused by phosphorylation
|
|
Computational prediction of NIPBL phosphorylation sites
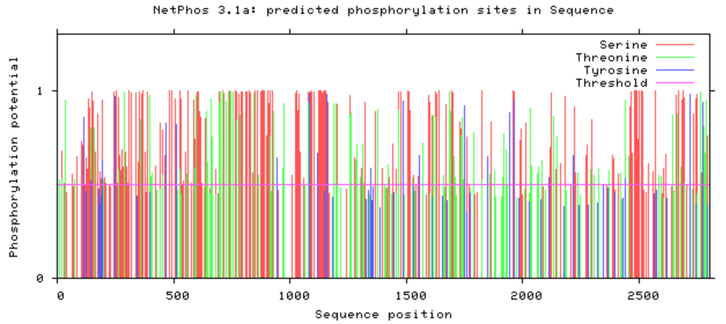
Online resources such as NetPhos are capable of predicting the phosphorylation sites on a protein given the peptide's sequence. Below is the NetPhos computational prediction for the human NIPBL protein sequence. The prediction indicates that NIPBL is more heavily phosphorylated on the N terminus of the protein, indicated by the phosphorylation potential seen between sequence positions 100 and 1100. This has potentially interesting applications as the N terminus of NIPBL is strongly conserved between species.
Online resources such as NetPhos are capable of predicting the phosphorylation sites on a protein given the peptide's sequence. Below is the NetPhos computational prediction for the human NIPBL protein sequence. The prediction indicates that NIPBL is more heavily phosphorylated on the N terminus of the protein, indicated by the phosphorylation potential seen between sequence positions 100 and 1100. This has potentially interesting applications as the N terminus of NIPBL is strongly conserved between species.
|
Colored bars represent the probability of the color specified amino acid to be phosporylated at that position.
|
Preparation for phosphoproteomics MS experiment
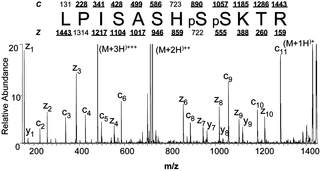
When a protein becomes phosphorylated it increases in mass because the phosphate group has some mass associated with it. Mass spectrometers can capture the mass difference in the molecular weight and then output data to be interpreted by bioinformatic algorithms. Using another ExPASy resource called "Compute pl/Mw" I computed the theoretical molecular weight of a non-phosphorylated NIPBL. I can now compare my theoretical weight to the weight of the phosphorylated peptides predicted by the bioinformatics algorithms and could manually estimate the phosphorylation.
MS spectrum pre-processing Theoretical mass to charge ration
When a protein becomes phosphorylated it increases in mass because the phosphate group has some mass associated with it. Mass spectrometers can capture the mass difference in the molecular weight and then output data to be interpreted by bioinformatic algorithms. Using another ExPASy resource called "Compute pl/Mw" I computed the theoretical molecular weight of a non-phosphorylated NIPBL. I can now compare my theoretical weight to the weight of the phosphorylated peptides predicted by the bioinformatics algorithms and could manually estimate the phosphorylation.
MS spectrum pre-processing Theoretical mass to charge ration
|

|
References
1.) What is proteomics? (2016, June 08). Retrieved April 12, 2018, from https://www.ebi.ac.uk/training/online/course/proteomics-introduction-ebi-resources/what-proteomics
2.) Koidakis, J., & Chatziharalambous, D. (2010). Mass Spectrometry: Structure Determination of Proteins and Peptides. Comprehensive Natural Products II,9, 457-496. doi:10.3897/bdj.4.e7720.figure2f
3.) Phosphorylation. (n.d.). Retrieved April 12, 2018, from https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/phosphorylation.html
1.) What is proteomics? (2016, June 08). Retrieved April 12, 2018, from https://www.ebi.ac.uk/training/online/course/proteomics-introduction-ebi-resources/what-proteomics
2.) Koidakis, J., & Chatziharalambous, D. (2010). Mass Spectrometry: Structure Determination of Proteins and Peptides. Comprehensive Natural Products II,9, 457-496. doi:10.3897/bdj.4.e7720.figure2f
3.) Phosphorylation. (n.d.). Retrieved April 12, 2018, from https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-biology-learning-center/protein-biology-resource-library/pierce-protein-methods/phosphorylation.html
