This webpage was produced as an assignment for Genetics 564, an undergraduate capstone course at UW-Madison.
Phylogeny
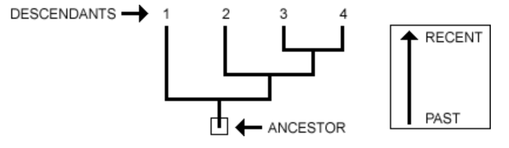
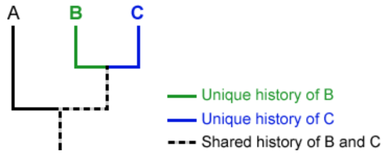
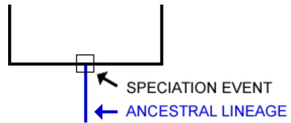
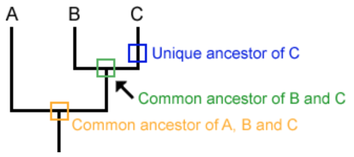
“A phylogeny, or evolutionary tree, represents the evolutionary relationships among a set of organisms or groups of organisms, called taxa (singular: taxon). The tips of the tree represent groups of descendent taxa (often species) and the nodes on the tree represent the common ancestors of those descendants.” [1.]
Interpreting phylogenetic trees
|
|
How are phylogenetic trees constructed?
|
Phylogenetics has come a long way since the time of Darwin. (See photo on right) Modern phylogenetics uses DNA sequences as opposed to physical characteristics to construct evolutionary trees. In fact, a plethora of algorithms exist for aligning genetic sequences and constructing trees. The basic process that these algorithms undergo is that they perform an alignment between two sequences and then continue performing alignments, the order in which these alignments are performed is what ultimately determines a tree's structure. These algorithms often differ in their assumptions about the data being used, their objectives, and in computational speed and accuracy.
|
|
Phylogeny of NIPBL
Here I have analyzed the evolution of the protein coding regions of NIPBL. To create these trees I utilized the ClustalW multiple sequence alignment algorithm and the Maximum Likelihood and Minimum Evolution tree construction algorithms. These computational tools have been collected into a neat little software package called MEGA, short for Molecular Evolutionary Genetics Analysis. As the evolutionary distance between organisms becomes smaller these two algorithms make different predictions. You can see this phenomenon happening because as you move up the tree the two algorithms made the same predictions until they reached the mammalian lineage and then their predictions diverge.
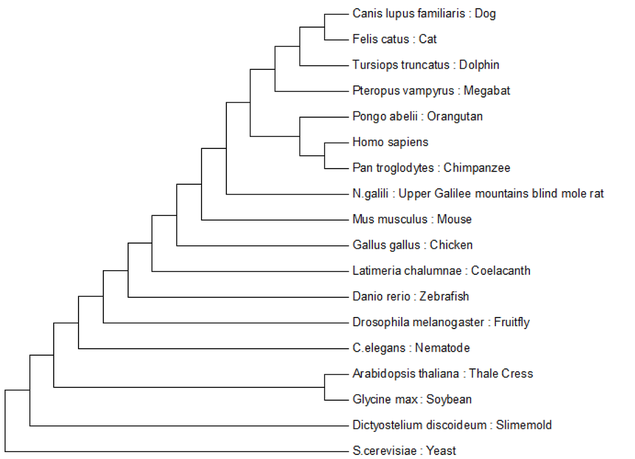
Minimum Evolution:
Tree construction method based on the assumption that the tree with the smallest sum of branch lengths is most likely to be the true one. [2.] |
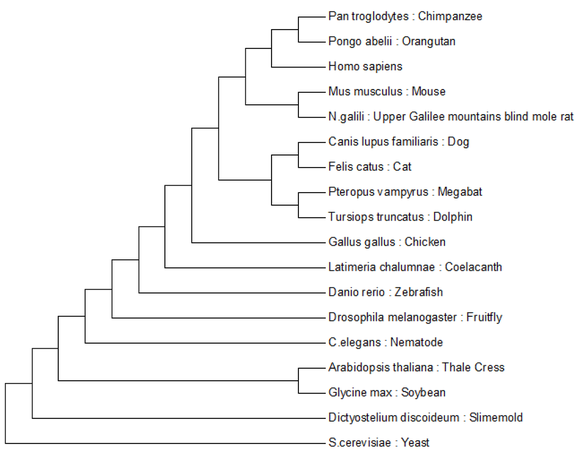
Maximum Likelihood:
Will consider all possible tree variations for the input sequences, chooses the tree that maximizes the probability of the input sequences given the tree structure. Utilizes a user defined statistical model to make these probability decisions. [3.] |
References:
1.) Reading Trees: A quick review. (n.d.). Retrieved March 15, 2018, from https://evolution.berkeley.edu/evolibrary/article/phylogenetics_02
2.) R., & Nei. (1993, September 01). Theoretical foundation of the minimum-evolution method of phylogenetic inference. | Molecular Biology and Evolution | Oxford Academic. Retrieved March 15, 2018, from https://academic.oup.com/mbe/article/10/5/1073/1037508
3.) Maximum Likelihood. (n.d.). Retrieved March 15, 2018, from https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
1.) Reading Trees: A quick review. (n.d.). Retrieved March 15, 2018, from https://evolution.berkeley.edu/evolibrary/article/phylogenetics_02
2.) R., & Nei. (1993, September 01). Theoretical foundation of the minimum-evolution method of phylogenetic inference. | Molecular Biology and Evolution | Oxford Academic. Retrieved March 15, 2018, from https://academic.oup.com/mbe/article/10/5/1073/1037508
3.) Maximum Likelihood. (n.d.). Retrieved March 15, 2018, from https://www.ncbi.nlm.nih.gov/Class/NAWBIS/Modules/Phylogenetics/phylo15.html
